Document Type : Original Article
Authors
- Emmanuel Israel Edache 1, 2
- Adumu Uzairu 2
- Paul Andrew Mamza 2
- Gideon Adamu Shallangwa 2
- Muhammad Tukur Ibrahim 2
1 Department of Pure and Applied Chemistry, University of Maiduguri, P.M.B. 1069, Maiduguri, Borno State, Nigeria
2 Department of Chemistry, Ahmadu Bello University, P.M.B. 1044, Zaria, Nigeria
Abstract
Coronavirus disease 2019 (COVID-19) is a pandemic disease caused by the SARS-coronavirus-2, which has a high rate of infection. Regardless of the advancements made in the creation of vaccines, it is urgently necessary to identify antiviral substances that can more effectively combat the SARS-coronavirus-2. The SARS-coronavirus-2 main protease is essential for viral transcription and replication. Using molecular docking, molecular dynamics simulation, and free binding energy calculations based on molecular mechanics/generalized Born surface area (MM/GBSA) approaches, an in silico technique was used in this study to help clarify the inhibitory potential of (N-(4-carbamoylphenyl)-8-cyclopropyl-7-(naphthalen-1-ylmethyl)-5-oxo-2,3-dihydro-[1,3]thiazolo[3,2-a]pyridine-3-carboxamide (aka, compound 36) against the main protease of SARS-coronavirus-2. Four software programs: AutoDockFR, AutoDock Vina v1.2.3, CABS-Flex2.0, and fastDRH servers were used to investigate the proteins binding sites, docked the ligands into the crystal structure of SARS-coronavrus-2 Mpro, check the stability of the complexes using molecular dynamics simulations, and MM/GBSA calculations, respectively. The standard drugs have all shown positive interactions with the main protease of the virus, but compound 36 has the highest negative binding affinity of them all. Computer-aided drug design was used to create a few compound 36 derivatives, and pharmacokinetic studies and molecular docking studies were conducted to assess their drug-like characteristics. These compounds (D3 and D6) have better binding affinities than the template and the conventional drugs. The top-scoring conformational complexes were subjected to molecular dynamics (MD) simulations in the following section of the study to further examine the complexes stability and the interactions between the ligand and receptor. The MM/GBSA further demonstrated that net free binding energies were primarily raised by Van der Waals interactions. The present investigation serves as a foundation for investigating the improved binding capacities and structural characteristics of SARS-CoV-2 Mpro variants to develop fresh anti-viral drugs.
Graphical Abstract
![Molecular Docking, Molecular Dynamics Simulation, and MM/GBSA Studies of (N-(4-Carbamoylphenyl)-8-cyclopropyl-7-(naphthalen-1-ylmethyl)-5-oxo-2,3-dihydro-[1,3]thiazolo[3,2-a]pyridine-3-carboxamide for Its Potential Use Against SARS-CoV-2](javascript:loadModal('Molecular Docking, Molecular Dynamics Simulation, ... ', 'data/jaoc/coversheet/701701492262.jpg'))
Keywords
- SARS-coronavirus-2 Mpro
- Docking simulations
- ADME
- Molecular dynamics simulations
- MM/GBSA calculations
Main Subjects
Introduction
The COVID-19 pandemic has become a serious threat to human health worldwide. The new coronavirus that causes COVID-19 is called SARS-CoV-2, or severe acute respiratory syndrome coronavirus [1]. It is an enveloped, positive-strand RNA virus that affects people and causes symptoms like fever, myalgia, coughing, headaches, dyspnea, and severe pneumonia [2]. There are dire social and economic repercussions from the current coronavirus pandemic [3]. The SARS coronavirus, or SARS-CoV, gained significant attention in coronavirus research following the significant outbreaks of severe acute respiratory syndrome (SARS) in 2003 and 2019 [4]. A serious public health concern since 2019 is the recently discovered coronavirus disease or COVID-19. The agent responsible for the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) [5] is evolving through multiple stages [2], and appears to be more lethal [1]. Important organs within the host body are being affected by the virus, which is spreading quickly through respiratory droplets [6]. In addition to directly causing tissue invasion, SARS-CoV-2 can also cause an overreaction of the host immune system, which often results in a cytokine storm and is a major cause of multiorgan dysfunction [7]. Over 635,709,158 people worldwide have been impacted by the terrible pandemic brought on by the SARS-coronavirus-2 virus. In the previous 26 months (i.e. 2019-2022), it caused the deaths of almost 7 million people [8]. As of November 23, 2022, the COVID-19 pandemic has caused 6,603,803 million deaths and 635,709,158 confirmed cases [9]. At the end of 2021, SARS-coronavirus-2 variants with distinct characteristics were discovered, which have facilitated the emergence of new waves of infections and the postponement of this pandemic [10]. As a result, several therapeutic targets are being suggested to combat SARS-coronavirus-2, which interacts with the protein spike glycoprotein and is crucial in the development of the disease [11]. All other enzymes, such as the main protein (M), an envelope protein (E), and a non-structural protein (Nsp), participate in the discharge of the viral genome into the host cell [12]. The main protease facilitates the viral fusion in the host membrane for the smoothening of cellular entry of the genetic material [13,14]. To stop the viral particles from entering the host cells, it has become more appealing to target the biochemical processes of the main protease [15]. As a result of recent research [16], it has been demonstrated that spike glycoprotein inhibition is more effective than that of SARS-2-S in preventing lung cell access. Consequently, creating therapeutic drugs that block spike glycoprotein function will have a positive effect on preventing future and present coronavirus occurrences. As computational chemistry can predict reaction processes in new experiments and the characteristics of unknown molecules, it allows new chemicals to be synthesized efficiently [17-22]. Given the importance of everyone's health, computational modeling has dominated further studies into the causes and prognoses of SARS-coronavirus diseases [15-16,23].
Docking simulation, molecular dynamic simulations (MDS), and molecular mechanics/generalized Born surface area (MM/GBSA) approaches are examples of computational methods that could provide answers to the significant issues that biomedical research alone might not be able to explain [24]. The goal of the current study is to develop stronger SARS-coronavirus inhibitors with improved binding infinity that may serve as more efficient COVID-19 inhibitors. To identify the crucial interactions and investigate the dynamic characteristics of ligands in the catalytic sites of the SARS-coronavirus-2 main protease, docking simulation, MDS, ADME, and MM/GBSA were carried out. The methodologies have shown to be a good potential route for drug design and would give valuable insights into the vital structural knowledge for continuing to synthesize the chemical antagonists.
Experimental
Methods
Source of dataset
The data set used in this work was taken from the PubChem database, as presented in Table 1. The 2D structures were sketched in Mavin View software and were cleaned using the clean structure option available in tools of Mavin View followed by energy minimized using minimization software Avogadro version 1.2.0. (https://Avogadro.c) using the steepest descent algorithm in the MMFF94 force field. PLIP was carried out using online freeware https://projects.biotec.tu-dresden.de/plip-web/plip/ DFT studies were performed using Gaussian 03 [17-22]. The main protease SARS-coronavirus-2, PDB id: 7CWL, and 6LU7 3D crystal structures were obtained from the protein data bank (https://www.rcsb.org/). Accordingly, the structures were modified to remove water molecules and the co-crystal ligands, and the PDB was updated to include any missing atom types using AutoDockTools v1.5.7 [25]. The protein was then changed into PDBQT and Kollman's charges were added with the aid of AutoDockTools v1.5.7.
In silico pharmacokinetics properties
The biodistribution characteristics of the chosen compounds were assessed using the pkCSM web server [26]. Using the drug-likeness rules, the pharmacokinetics and physicochemical properties were evaluated for the analysis [27-30].
Valuation of ligand interactions with SARS-coronavirus-2 Mpro
Using the molecular docking method and the EasyDockVina v2.2 software [31], the interactions of ligands with their corresponding receptor proteins were investigated. In AutoDockFR v1.2 [32], the grid was fixed around the active site, and the grids were predicted using the compute pocket (AutoSite 1.1) implemented. EasyDockVina v2.2, which incorporates AutoDock-Vina v1.2.3 [33,34], was used to finish studies on molecular docking simulation.
Grid dimensions
The center dimensions (x = 118.500, y = 107.250, and z = 168.750 Å) for PDB id 7CWL and PDB id 6LU7 (x = 60.000, y = 75.750, and z = 67.500 Å), respectively were measured in the present study. The grid box size dimensions (x, y, and z) were 186.043, 201.934, and 223.221 for PDB id 7CWL and -26.053, 13.285, and 58.250 for PDB id 6LU7. The spacing for both proteins was determined by 0.375 Å [32].
Molecular dynamics (MD) simulations
The CABS-Flex 2.0 server, which relies on coarse-grained protein movements, was used to do MD simulations of the top three complexes and the receptor (SARS-CoV-2 PDB: 6LU7) [35]. There were more than 50 cycles and 50 trajectory frames in 10 ns each, along with some extra distance restrictions like a global weight of 1.0. The complexes mobility was expressed using root-mean-square fluctuations (RMSF). In the default mode, the CABS-flex uses a set of distance restraints and simulation settings defined in the work of Kuriata and his coworkers [32] (Consistent View of Protein Fluctuations from All-Atom Molecular Dynamics and Coarse-Grained Dynamics with Knowledge-Based Force-Field).
Fast DRH server
Molecular mechanics (MM) of Poisson-Boltzmann surface area (PB) and Generalized Born Surface Area (GBSA) (MM/PB(GB) SA) techniques are used to determine the rescoring docking poses for protein-ligand inhibitors. This freely accessible online web server may be found at https://cadd.zju.edu.cn/fastdrh/. These techniques are used to estimate the binding affinity of the chosen compounds.
Results and Discussion
Practical screening of SARS-coronavirus-2 main protease target
The highest-scoring substances that could inhibit SARS-coronavirus-2 main protease (Mpro) (PDB code: 7CWL and 6LU7) were found through virtual screening. Molecular docking was also used to compare some well- known medications that are currently being researched for their potential role in the treatment of SARS-coronavirus-2. Nine of the highest-scoring docked poses for each compound have been found through molecular docking calculations. Table 1 lists the highest docking scores of each compound. Results revealed that the docking scores of the investigated compound 36 “(N-(4-carbamoylphenyl)-8-cyclopropyl-7-(naphthalen-1-ylmethyl)-5-oxo-2,3-dihydro-[1,3]thiazolo[3,2-a]pyridine-3-carboxamide” has the least negative binding affinity than those of the reference or standard drugs. The standard drugs interacted within the Mpro active site, establishing their potential strength in barricading the vital site in SARS-coronavirus-2 Mpro (Figure 1).
Figures 1A and 1B showed the Hydroxychloroquine interactions against 7CWL and 6LU7 receptors with the binding affinity of -5.939 and -4.989 kcal/mol, respectively. Lopinavir with binding affinity -7.912 and -8.22 kcal/mol are presented in Figure 1C and 1D. Figures 1E and 1F of Ritonavir interactions have a binding affinity of -7.266 and -6.595 kcal/mol with the receptors. Ruxolitnib with binding affinity -6.509 kcal/mol against the 7CWL receptor and -7.075 kcal/mol against the 6LU7 receptor is presented in Figure 1G and 1H, respectively. Azithromycin binding affinity against the two receptors is -7.785 and -6.841 kcal/mol. The binding interactions are displayed in Figure 1I and 1J, respectively. Among the common medications, Lopinavir has one of the highest binding affinities to 7CWL due to the presence of three regular hydrogen bonds with Ser373, Asn343, and TRP436 at distances of 4.53 Å, 3.92 Å, and 5.23 Å respectively. The complex formed is stabilized by two carbon-hydrogen bonds with ASN439 and ASN440 at a distance of 5.29 Å and 4.78 Å, which produces a potent cohesive environment. The Lopinavir is also stabilized by hydrophobic interactions on the two farthest ends of the molecule. On one end, the dimethphenyl group is making amide-pi stacking interactions with ASN343 and PHE338 at a distance of 4.62 Å and 6.38 Å as is shown in the 2D analyses (Figure 1C). The analysis of Lopinavir interactions (Figure 1D) revealed that the 2,6-dimethphenyl group was oriented to the N-terminus of the receptor virus, while the phenolic group was directed to the C-terminus (VAL104). Furthermore, the 2,6-dimethphenyl group was bound in a hydrophobic pocket consisting of residue VAL104. Specifically, the phenolic group of the ligand form three hydrophobic interactions consisting of the residues PRO293 (4.99 Å), VAL202 (6.67 Å), and ILE249 (4.68 Å), respectively. The N-H group of the ligand forms unfavorable donor-donor interaction with the amino acid GLU110 of the 6LU7 receptor, as displayed in Figure 1D.
Table 1. The affinities of the selected compounds against SARS-CoV-2 main protease
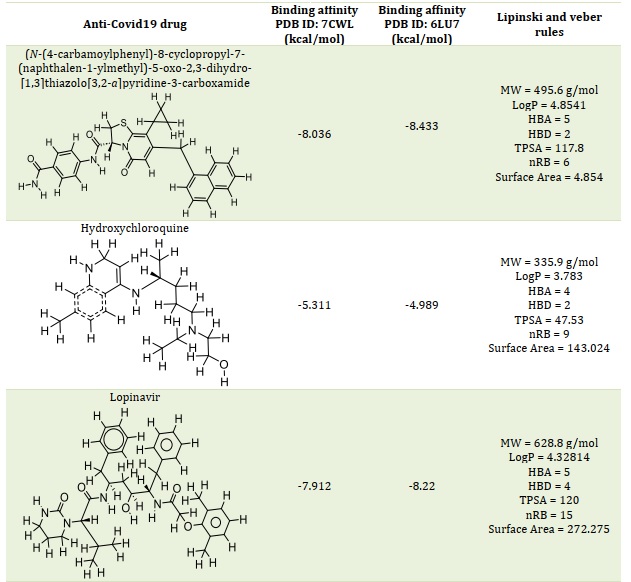
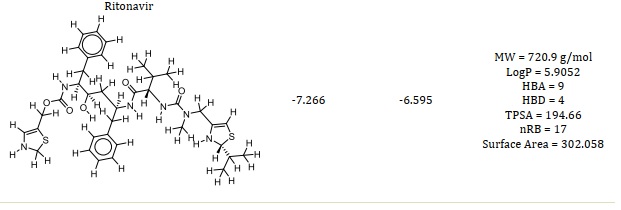
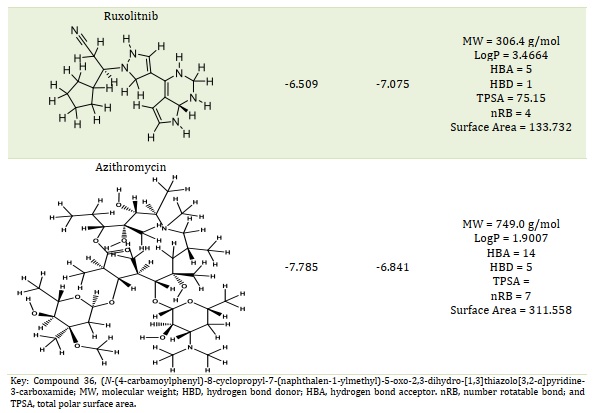
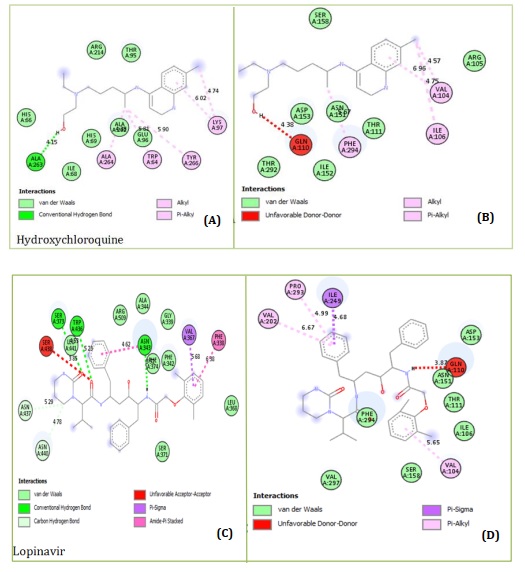
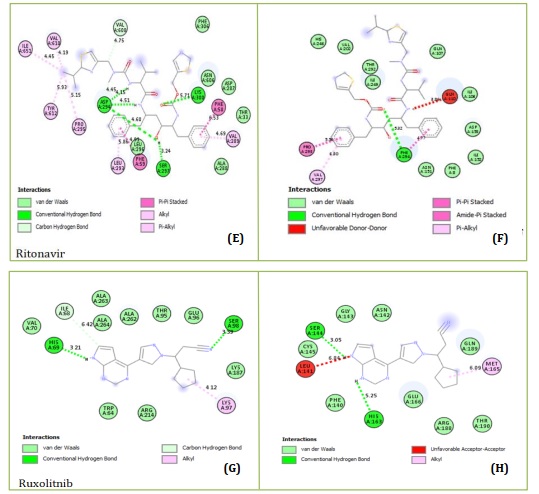
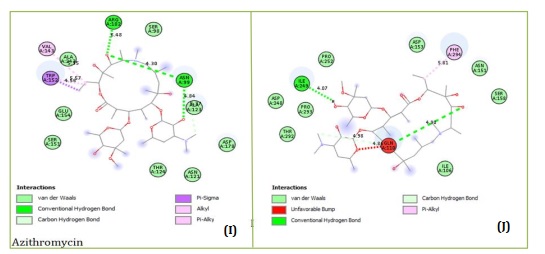
Figure 1. Key residues and two-dimensional interactions in the inhibitory binding between 7CWL and the approved standard medications
The findings also indicate that compound 36, which forms numerous hydrophobic interactions, van der Waals contacts, and one conventional hydrogen bond with the Mpro of the SARS-coronavirus-2 7CWL and 6LU7 receptors, respectively, is the most promising ligand. Most of the binding site is situated in a hydrophobic cavity that is surrounded by the amino acids PHE220, VAL289, PHE59, and PHE58. They are nine amino acids with are GLN218, THR33, ALA288, ASP290, SER297, LEU293, LEU296, LYS300, and ASP294 are involved in Van der Waals interactions. There are five hydrophobic interactions: two pi-pi stacked interactions with PHE220 at 6.36 Å and 6.82 Å, one pi-pi T-shaped interaction with PHE59 at 4.52 Å, one alkyl and one pi-alkyl interaction with VAL289 (4.54 Å) and PHE58 (6.70 Å), respectively. The conventional hydrogen bond was observed with amino acid ASN30 with a distance of 5.74 Å, as presented in Figure 2. Van der Waals interactions, which helped forge a potent performance and sustainability and stabilize the complex formed, are also associated with the binding affinity of compound 36 with the 6LU7 receptor (Figure 3).
Compound 36 forms four conventional hydrogen bonds with CYS145 at a distance of 5.39 Å and 4.02 Å, HIS41 (5.01 Å), and THR26 (4.02 Å) and two hydrophobic interactions MET165 and CYC145 with Mpro 6LU7 and yields the binding affinity -8.433 kcal/mol by docking.

Figure 2. 3D and 2D fundamental interaction of compound 36 with the protein PDB id: 7CWL
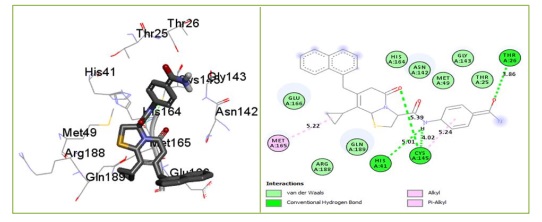
Figure 3. 3D and 2D fundamental interaction of compound 36 with the protein PDB id: 6LU7
The classification of a compound as a potential drug candidate must adhere to some rules and models. The Lipinski Rule of Five is the one that is most widely accepted [34, 35]. One of the biggest obstacles to the marketing of medications is the dearth of pharmacokinetic research. As a result, the study was also planned to assess the pharmacokinetic, physicochemical, and drug-likeness characteristics of the six chosen compounds using computational techniques. Lead compounds' permeability, promiscuity, potency, solubility, and selectivity are all influenced by lipophilicity [36, 37]. Except for compound 36, hydroxychloroquine, and rufinib, all of which have molecular weights within the permitted range (MW < 500), whereas azithromycin, lopinavir, and ritonavir have molecular weights larger than 500 g/mol. In contrast to lopinavir, ritonavir, and azithromycin, this suggested that compound 36 and hydroxychloroquine with MW ≤ 500 g/mol can be readily absorbed, dispersed, and transported. According to the Rule of Five (Ro5) [27-29], drug-like compounds should have a certain number of hydrogen bond donors (nHBD) ≤ 5 and acceptors (nHBA) ≤ 10. Except for azithromycin, all examined drugs' nHBA and nHBD were found to be within Lipinski's limit range (Table 1). This suggests that when administered, the substances may be well absorbed or permeable from the gastrointestinal tract [38]. One of the often-employed filters in the drug discovery process is the number of rotatable bonds, which is a measure of molecular flexibility [39]. Each compound's number of rotatable bonds was examined in this investigation, and the findings are presented in Table 1. The compounds Ritonavir (nRB = 17), Lopinavir (nRB = 15), Hydroxychloroquine (nRB = 9), Azithromycin (nRB = 7), Compound 36 (nRB = 6), and Ruxolitnib (nRB = 4) were found to have the most rotatable bonds. Good bioavailability compounds have at least fifteen rotatable bonds. Compounds 36, Ruxolitnib, Azithromycin, and Hydroxychloroquine all fell into the acceptable range (nRB ≤ 15), suggesting that they may be permeable and orally bioavailable. Other compounds under examination exhibited low oral bioavailability and greater flexibility due to a high number of rotatable bonds (15 ≤ nRB). Table 1 indicates the compounds' logP values. Ritonavir deviated from the Ro5 (logP > 5), while all other drugs under investigation complied with it (logP < 5). Compounds with a high rate of rapid metabolic turnover, low solubility, and poor absorption are often lipophilicity (logP > 5). Furthermore, chemicals that are more lipophilic (logP > 5) are more likely to bind to hydrophobic protein targets other than the intended ones, which can have harmful consequences on biological systems.
Evaluation of drug likeness
An ADMET analysis of the reference and investigated medicines was performed using the pkCSM online server (Table 2). When compared to the two reference medications, compound 36 showed a higher human intestinal absorption (HIA) (100%), particularly to azithromycin (45.808%), suggesting poor absorption from the intestines following oral intake. The permeability of the blood-brain barrier (BBB) was evaluated concerning distribution analysis. For medications to function in the central nervous system (CNS), the blood-brain barrier (BBB) should be permeable to facilitate the drugs' absorption from the bloodstream into the brain. On the other hand, the brain absorption of non-CNS-active medicines may result in CNS toxicity. Ritonavir (-1.665), Lopinavir (-0.83), and azithromycin (-1.857) were projected to have the lowest BBB permeability. According to Elekofehinti et al. [40], the BBB values for the remaining chemicals varied from -0.574 to 0.074, suggesting a modest chance of crossing the BBB. Concerning the examination of metabolism, the CYP3A4 parameter was evaluated. According to Edace et al. [23,41], CYP3A4 is a significant isoenzyme that participates in the oxidative biotransformation and metabolism of about 60% of medications and xenobiotics in humans. The compounds are predicted to be substrates for the CYP3A4 isoenzyme, indicating that they are properly metabolized. In contrast, CYP3A4 was predicted to be inhibited by some of the compounds (compound 36, lopinavir, and ritonavir). CYP3A4 inhibition causes drug-drug interactions (DDI) and associated toxicity, which is caused by the bioaccumulation of co-administered medications [42]. As a result, Lopinavir binding affinity is somewhat limited by its incompatibility with metabolic processes. Compounds Ruxolitnib, Lopinavir, Ritonavir, and Hydroxychloroquine all had more excretion through the hepatic and renal pathways, as seen by their higher total clearance values (0.459-1.152) ml/min/kg compared to compound 36 (-0.634 ml/min/kg) and Azithromycin (-0.424 ml/min/kg). The body will hold onto compound 36 for a longer amount of time if its total clearance value is low. Ames toxicity test results for all reference medications were negative, indicating that they are non-mutagenic, the exceptions being compound 36 and hydroxychloroquine. Compound 36 and hydroxychloroquine tested positive in the Ames toxicity test, indicating that they may be carcinogenic and mutagenic [24, 38].
New compounds design and structure-based predictions
Compound 36 was utilized as a template to change its molecular structure based on the determined molecular docking scores and associated analytical outcomes. The binding affinity of the compounds under study is found to be significantly influenced by the hydrogen bond donor, hydrogen bond acceptor, and a few critical hydrophobic groups. Ten additional molecules are suggested in this study, and they have all undergone the same optimization processes as the reference and chosen compounds. Table 3 displays the expected binding affinities and chemical structures of the developed compounds against the two receptors (PDB: 6LU7 and 7CWL). In comparison to the most powerful molecule (36), the newly developed compounds have a high binding affinity.
Table 2. ADMET prediction of the six selected compounds

Table 3. The proposed drugs' molecular binding affinities against SARS-CoV-2 Mpro
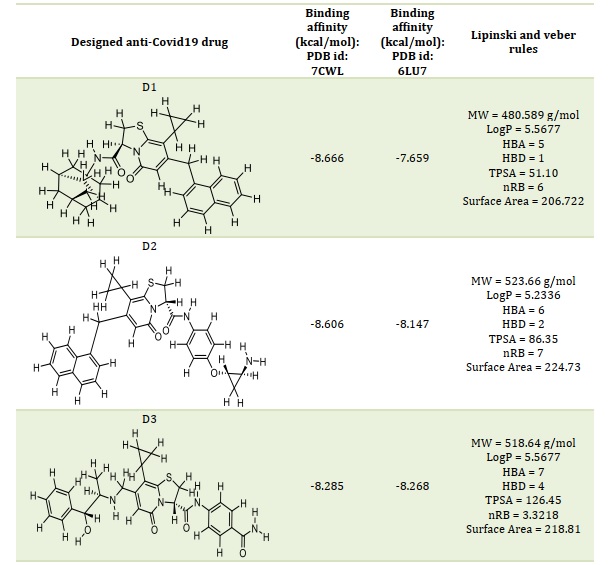

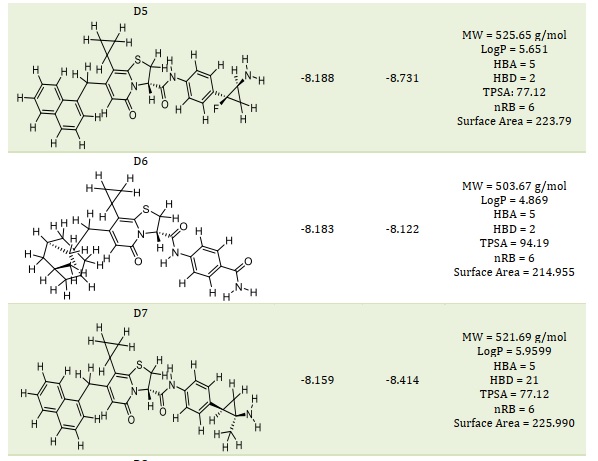

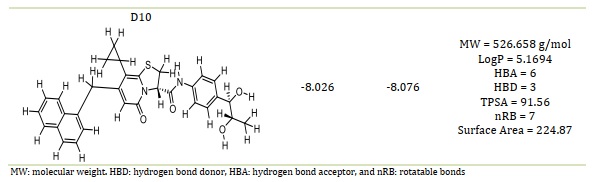
Table 4. The smiles code structure of the designed compounds

Table 4 presents the smile code structure of the created chemicals. In comparison to the template and standard medicines, the compounds D1 through D10 (Table 2) showed a greater binding affinity and a positive binding interaction when they were deeply docked inside the binding site of the SARS-CoV-2 MPro. The findings of the molecular docking analysis lend credence to the template's potential for further development as a new coronavirus (SARS-CoV-2) inhibitor.
Molecular docking of the designed compound
Based on molecular docking, the top 10 proposed compounds had binding affinities against SARS-CoV-2 Mpro ranked between -8.666 and -8.026 kcal/mol for PDB id 7CWL and -8.731 and -7.556 kcal/mol for PDB id 6LU7 receptors, respectively (Table 2). It is noteworthy that the bulk of them are members of the template structure's bicyclic 2-pyridone family, which is a well-known as N-(4-carbamoylphenyl)-8-cyclopropyl-7-(naphthalen-1-ylmethyl)-5-oxo-2,3-dihydro-[1,3]thiazolo[3,2-a]pyridine-3-carboxamide (aka, compound 36). The most effective putative inhibitors of SARS-CoV-2 Mpro were the proposed compounds D1 and D5, which had binding affinities of -8.666 and -8.731 kcal/mol for PDB ids 7CWL and 6LU7, respectively. Furthermore, it was found that several of the proposed compounds violated Lipinski's rule of five, at least in terms of molecular weight (Table 2). Nevertheless, since these designed compounds outperform existing medications on the market and have template binding affinity, their physicochemical should not be undervalued. To determine the precise interactions between the top two developed compounds and SARS-CoV-2 Mpro, the Discovery Studio program was used. One pi-donor hydrogen bond against the SARS-CoV-2 Mpro 7CWL receptor was revealed to have formed by the D1 complex via PHE898. However, by forming an electrostatic contact with the receptor, ASP796 helped to contribute to the robust binding. Furthermore, the receptor's hydrophobic contacts were formed by residues PRO897, ALA893, and TYR789 (Figure 4A). Only hydrophobic contacts are formed with the receptor by the proposed compound D1, which is at the 8th position and has a binding affinity of -7.659 kcal/mol against SARS-CoV-2 Mpro PDB id 6LU7, as shown in Figure 4B. The developed compound's binding affinity is superior to those of typical medications (Ruxolitnib, Azithromycin, Ritonavir, and Hydroxychloroquine), except Lopinavir, which has a binding affinity of -8.22 kcal/mol with the 6LU7 receptor. Nevertheless, this should not be taken as an indication of the compound's suppression. At the fifth position, the proposed chemical D5 makes two conventional hydrogen bonds with LEU176 at 4.26 Å and 5.36 Å, respectively, with a binding affinity of -8.188 kcal/mol against the SARS-CoV-2 Mpro PDB id 7CWL receptor. The compound forms one halogen interaction with ASN99 with six hydrophobic interactions with VAL143, ALA243, ALA123, HIS245, and TRP152, respectively, as presented in Figure 5A. The designed compound D5 against the 6LU7 receptor at position one with a binding affinity of -8.731 kcal/mol, forms one carbon-hydrogen bond interaction with PHE249 at a distance of 4.33 Å. One halogen interaction with PRO293 and three hydrophobic interactions with PHE249 and ILE106, as displaced in Figure 5B.
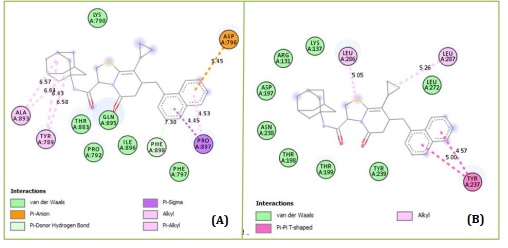
Figure 4. Protein-ligand interactions of D1 with (A) 7CWL and (B) 6LU7 as predicted by Discovery Studio
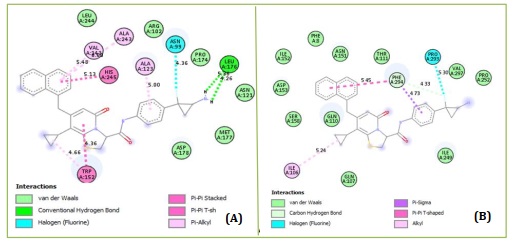
Figure 5. Protein-ligand interactions of D5 with (A) 7CWL and (B) 6LU7 as predicted by Discovery Studio
ADMET prediction of the designed compounds
The developed compounds demonstrated high levels of human intestinal absorption in the range of 81.263% to 100%, and they were labeled as being completely absorbed by the human enteric. In contrast to plasma, compounds with logVDss < -0.15 are thought to be poorly dispersed in tissues. Except for the intended designed compound D6, the majority of the rest of the designed compounds were distributed in the plasma, according to Table 5's logVDss data. All of the created compounds were anticipated to be CYP450 3A4 subtype substrates for metabolism, suggesting that CYP 3A4 may be able to properly metabolize them. Furthermore, all of the created compounds aside from D6 could inhibit the 2C9 subtype, but none could inhibit the 2D6 subtype. However, one or more of the intended chemicals may inhibit some of the cytochrome P450 subtypes. Except for proposed compounds D1 and D6, all designed compounds could be filtered by renal and hepatic tissues in combination due to the high values of total clearance. The expected toxicity showed that the designed compounds D3 and D6 were not mutagenic, demonstrating the molecules' safety crucial factor in the development of an effective medication. The created compounds (D3 and D6) showed encouraging outcomes from toxicity and computational pharmacokinetic analyses, suggesting that they could be useful inhibitors of SARSCoV-2 for future research. Comparing the developed compounds D3 and D6 against template and conventional medicines, it demonstrated a reasonably decent ADMET profile and good potential as a SARS-CoV-2 inhibitor.
Table 5. The ten chosen designed compounds' ADMET characteristics

The pharmacokinetic compliance of a proposed chemical (D1 to D10) is taken into consideration when determining the ADMET. Molecular dynamics simulations employ just those substances that meet these variations without default. D3 and D6 are the best-designed compounds as a result, notably in terms of AMES toxicity. The designed compound docking data indicate that D3 has interacted through five conventional hydrogen bonds with THR791 (3.74 Å), LYS795 (5.48 Å and 6.86 Å), SER803 (3.63 Å), and PHE802 (5.20 Å) with the active region of the PDB 7CWL receptor. It interacts with LYS795 just once electrostatically, at a distance of 6.86 Å, and it interacts with PRO809, LEU805, PRO807, ILE794, PHE797, and LYS795 six times hydrophobically, in that order. Finally, as demonstrated in Figure 5-2A, it creates an unfavorable donor-donor relationship with SER875. Three conventional hydrogen bonds with THR292 (3.46 Å), ASN151 (3.68 Å), and ARG105 (6.19 Å), one carbon-hydrogen bond with SER158 (4.17 Å), one hydrophobic interaction with VAL104, and an unfavorable donor-donor interaction with THR111 were found to have been the modes of interaction between D3 and the PDB 6LU7 receptor's active site in Figure 5-2B. The PDB 7CWL receptor on the proposed molecule D6 exhibited three hydrophobic interactions with TYR789, PRO792, and PRO897. In addition, it produced one pi-donor and three conventional hydrogen bonds, which were ALA899 (3.58 Å), PHE898 (4.74 Å), ASP796 (3.76 Å), and PHE898 (6.18 Å), respectively. Furthermore, electrostatic contact was discovered at location ASP796 in Figure 6A, at a distance of 5.15 Å.
The PDB 6LU7 receptor-containing designed chemical D6 interacted with CYS145 at a distance of 4.81 Å, THR26 at a distance of 3.01 Å, and ASN142 at a distance of 3.92 Å in two conventional hydrogen bonds and carbon-hydrogen bond interactions. Together with PRO168, it also forms one hydrophobic alkyl interaction. As seen in Figure 6B, the benzoic ring of D6 forms a pi-sulfur contact with CYS142. When compared to typical medications for the investigated enzymes, the binding affinity of the proposed chemical in SARS-CoV-2 MPro demonstrates a greater number and variety of positive interactions. Hydrophobic contacts, electrostatic interactions, and traditional hydrogen bonding may affect the protease's structure and activity and be crucial in maintaining the complex shape. Comparing the created compounds D3 and D6 to the template structure (CID: 12328833, also known as compound 36) and the reference medications, the designed compounds were able to dock deeply into the binding site of the SARS-CoV-2 MPro.
This resulted in a favorable binding interaction and a higher binding affinity. The proposed compounds D3 and D6 have the potential to be further developed as a new inhibitor of coronavirus (SARS-CoV-2) as supported by the findings of the molecular docking investigation.

Figure 5-2. Designed compound D3 and the active site in the (A) 7CWL and (B) 6LU7 crystal complex
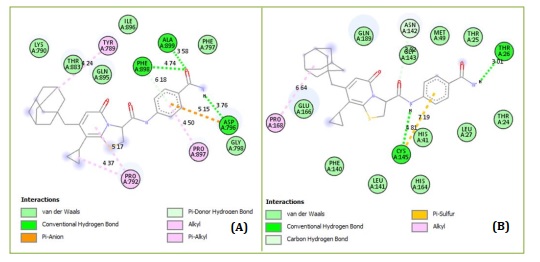
Figure 6. Designed compound D6 and the active site in the (A) 7CWL and (B) 6LU7 crystal complex
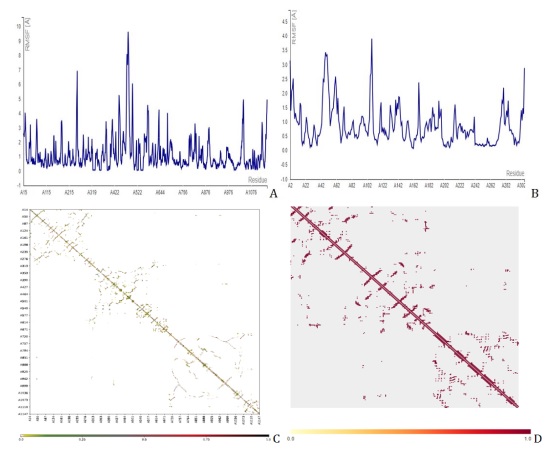
Figure 7. Molecular dynamics simulation showing the fluctuation plot of compound 36 with (A) PDB id 7CWL, (B) PDB id 6LU7, (C) contact map of 7CWL, and (D) contact map of 6LU7
Molecular Dynamics (MD) simulations analysis
With all other parameters set to default, the CABS Flex 2.0 web server was used to present the root-mean-square fluctuation (RMSF) and contact frequency for compound 36, Figure 8 for D3, and Figure 9 for D6, which further validated the molecular docking results. The coarse-grained protein modeling provided information on the flexibility and rigidity of the protein-ligand complex. The average movement of individual atoms or groups of atoms with the matching structure (configuration) is measured by the RMSF. Compounds D3 and D6 complexes that were constructed were shown in their contact maps, which overlapped the configuration and connection map of the tenth models of versatile calculation of the two receptors (7CWL and 6LU7) to the template structure compound 36. To determine whether a structure is reliable over the computation period (simulation) or if it differs from the early coordinates (initial). The RMSF was used to infer the amino acid sequence of the active site. The receptors to the catalytic residues of the ligands combination were all stable, according to the RMSF. This implies that the SARS-CoV-2 Mpro receptors have more stiff conformations that are appropriate for binding relationships. By measuring the relative movements of the residues, the cross-correlation analysis (contact map) may illustrate the interactions between residues and between different areas. Figures 7C and 7D for compound 36, Figures 8C and 8D for D3, and Figures 9C and 9D for D6 show the normalized correlation, which ranged from -1.0 to +1.0 (dark blue to white to red). Residues that migrated in the same direction were indicated by positive correlations (red patches) while residues that traveled in the opposite direction were indicated by negative correlations (blue regions). A higher positive correlation or negative correlation was represented by the darker color. A low correction was deemed to be the white areas between -0.25 and 0.25. The diagonal elements exhibit the highest correlation because each diagonal point represents the Cα atom of the same residue along both axes. These results showed that the compound D3 and D6 constructs we generated, along with the template structure, are acceptable for processing. We saw the least amount of residue variations across the board for every compound. It was discovered that the RMSF variation was less than 10 Å for every complex. As a result, it was discovered that every protein-ligand combination examined was stable under physiological conditions.
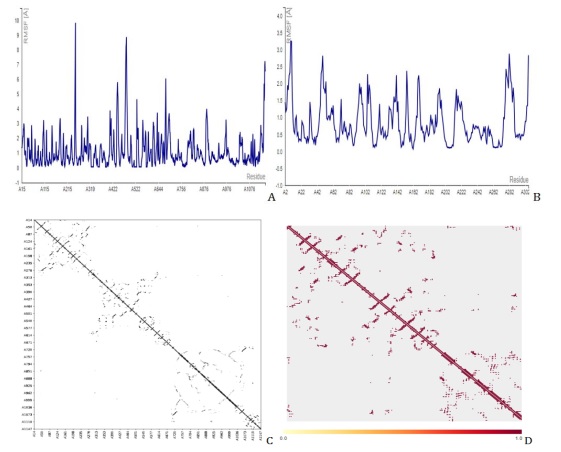
Figure 8. Molecular dynamics simulation showing the fluctuation plot of D3 with (A) PDB id 7CWL, (B) PDB id 6LU7, (C) contact map of 7CWL, and (D) contact map of 6L
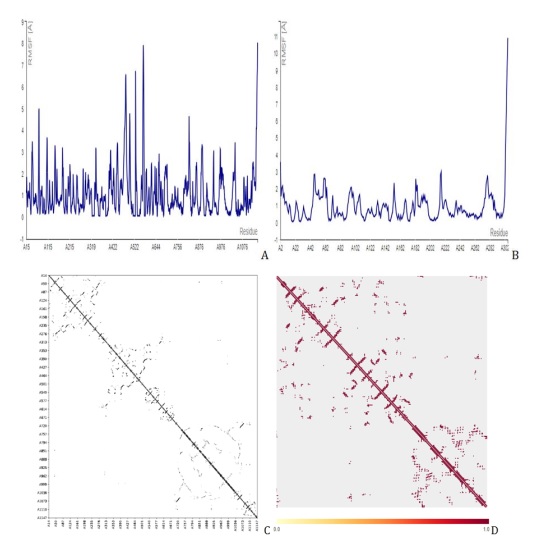
Figure 9. Molecular dynamics simulation showing the fluctuation plot of D6 with (A) PDB id 7CWL, (B) PDB id 6LU7, (C) contact map of 7CWL, and (D) contact map of 6LU7.
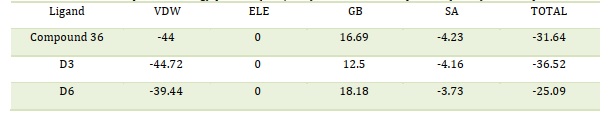
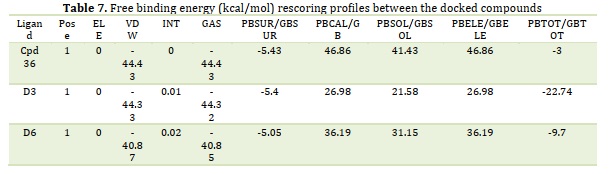
By providing the cartesian coordinates of the relevant receptor and ligand molecules, fastDRH, an online web service, was able to determine the hotspot-free energy for each of the three complexes. For hotspot-free energy calculation, we have used the PB3 (radii = parse, γ = 0.00542, and β = 0.92) MM/PBSA procedure based on the pose ranking. Table 6 provides a summary of the hotspot score energies and energy terms for each of the three complexes. The hotspot score between the ligand and the receptor molecule was found to be greater in all three complexes, as presented in Table 6. The D3 complex has a comparatively greater binding score energy than the other two complexes. MM/GBSA approaches have shown to be much more effective in rescoring operations after docking analysis [34]. MM-PB (GB)SA approaches are thought to be a great option for determining appropriate binding structures and calculating binding energies due to their effective performance in scoring procedures [15-16,23-24]. By applying the MM/GBSA method, fast DRH can predict accurate binding conformations as well as the precise binding energy profile of complexes. Table 7 shows the MM/PB(GB)SA findings of the SARS-CoV-2 Mpro PDB 6LU7 protein that was calculated using the rapid DRH method. The SARS-CoV-2 Mpro decomposition energy consisted of the following: Van der Waals contribution (VDW), electrostatic energy (ELE), total gas phase energy (TGAS), non-polar and polar contribution to solvation (TGBSOL), and final estimated binding free energy (TGBTOT). The template complex compound 36 containing SARS-CoV-2 PDB 6LU7 had the lowest binding score of -3 kcal/mol in the current investigation. Table 7 provides the profiles of free binding energies (kcal/mol) rescoring between drug 36 and the docked 6LU7 receptor. The D3 complex including the receptor displayed the greatest binding score of -22.74 kcal/mol, whereas the D6 complex displayed a free binding energy score of -9.7 kcal/mol. MM/GBSA has shown very successful in emphasizing the significant residues on the binding surface of protein-ligand complexes concerning per-residue energy breakdown [24]. It is important to note that the majority of the interaction energy between ligands and proteins is derived from the combined energies of the VDW and the GAS phases. Electrostatic (ELE) and internal (INT) energies give the final bit of binding free energy. The results obtained validate the free energy of binding and the ligands propensity to attach to the protein structures, indicating their potential as study targets.
Table 6. The hotspot free energy profiles (kcal/mol) between docked protein (6LU7) and compounds
Conclusion
In silico methods were used in this study to look into compound 36's potential as an inhibitor of the SARS-coronavirus-2 main protease (Mpro). The findings of our study suggest that (N-(4-carbamoylphenyl)-8-cyclopropyl-7-(naphthalen-1-ylmethyl)-5-oxo-2,3-dihydro-[1,3]thiazolo[3,2-a]pyridine-3-carboxamide (aka, compound 36) and the designed compounds D3 and D6 could be developed as potential inhibitors that can target main protease (Mpro) proteins. Lipinski’s rule is important to determine the ADMET properties of bioactive and in the study, Lopinavir, Ritonavir, and Azithromycin did not obey the rule of five (RO5). Compound 36's bioavailability is in fair agreement with the Lipinski and Veber rule. Utilizing compound 36 as the template, a computer-aided design was performed to create SARS-CoV-2 Mpro inhibitors. AutoDock Vina v1.2.3 revealed the top binding pocket in the main protease protein (PDB id: 7CWL and 6LU7) along with their accessible surface area and volume. The binding affinities of the newly developed compounds were better than those of the reference medications and the template most especially compounds D3 and D6. The newly developed compounds exhibited improved pharmacokinetic profiles and binding energies of interactions than the reference medications due to their increased intestinal absorption in humans, as well as their ability to function as CYP-3A4 substrates and inhibitors and to demonstrate no toxicity from AMES. fastDRH calculated the hotspot free energy profiles of the protein-ligand complexes with the protein (PDB id: 6LU7) complex with the highest affinity profile scores. The insights about MM/GBSA were provided by fastDRH. The results of a molecular dynamics simulation showed that newly designed compounds (D3 and D6), docked there with significant binding free energies of -22.74 and -9.7 kcal/mol, respectively, were stable within the active site of Mpro of the SARS-coronavirus-2. Based on these results, the newly designed compounds can be tested against in vitro SARS-coronavirus-2 Mpro to advance the development of a secure and efficient treatment for SARS-coronavirus-2 infection.
Acknowledgments
The authors gratefully acknowledged the technical effort of Dr. S.N. Adawara, Department of Pure and Applied Chemistry, Faculty of Physical Science, University of Maiduguri, P.M.B. 1069, Maiduguri, Borno State, Nigeria.
Conflict of interest
The authors confirm that there are no known conflicts of interest in this study.
Statement of ethics
In this study, no method requiring the permission of the “Ethics Committee” was used.
Availability of data and materials
All data generated or analyzed during this study are included in this published article.
Orcid
Emmanuel Israel Edache: https://orcid.org/0000-0002-5485-0583
Adamu Uzairu: https://orcid.org/0000-0002-6973-6361
Paul Andrew Mamza: https://orcid.org/0000-0002-7177-8202
Gideon Adamu Shallangwa: https://orcid.org/0000-0002-0700-9898
Muhammad Tukur Ibrahim: https://orcid.org/0000-0002-6146-5667
Citation: E.I. Edache*, A. Uzairu, P.A. Mamza, G.A. Shallangwa, M.T. Ibrahim Molecular Docking, Molecular Dynamics Simulation, and MM/GBSA Studies on (N-(4-Carbamoylphenyl)-8-Cyclopropyl-7-(Naphthalen-1-Ylmethyl)-5-Oxo-2,3-Dihydro-[1,3]thiazolo[3,2-a]Pyridine-3-Carboxamide for Its Potential Use against SARS-CoV-2. J. Appl. Organomet. Chem., 2023, 3(4), 321-345.
)